

According to the World Health Organization, each year there are an estimated 1 billion cases of influenza, between 3-5 million severe cases and up to 650,000 influenza-related respiratory deaths globally. Seasonal flu vaccines must be reformulated each year to match the predominantly circulating strains. When the vaccine matches the predominant strain, it is very effective; however, when it does not match, it may offer little protection.
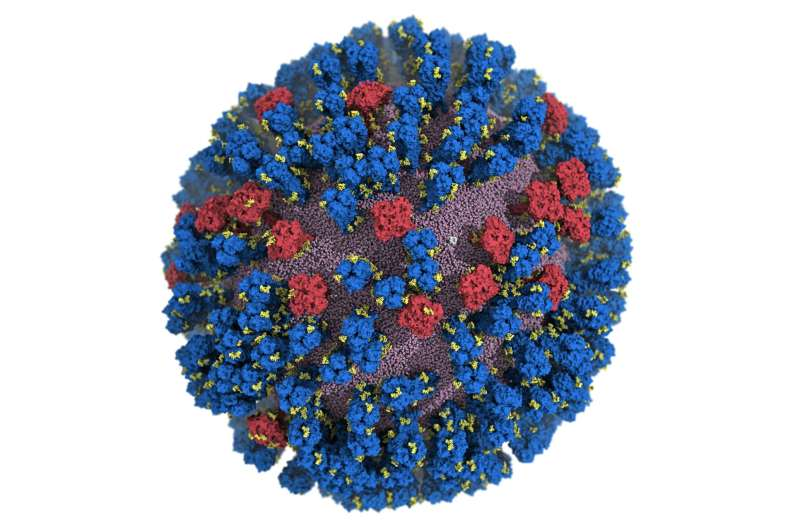
The main targets of the flu vaccine are two surface glycoproteins, hemagglutinin (HA) and neuraminidase (NA). While the HA protein helps the virus bind to the host cell, the NA protein acts like scissors to cut the HA away from the cell membrane allowing the virus to replicate. Although the properties of both glycoproteins have been studied previously, a complete understanding of their movement does not exist.
For the first time, researchers at the University of California San Diego have created an atomic-level computer model of the H1N1 virus that reveals new vulnerabilities through glycoprotein “breathing” and “tilting” movements. This work, published in ACS Central Science, suggests possible strategies for the design of future vaccines and antivirals against influenza.
“When we first saw how dynamic these glycoproteins were, the large degree of breathing and tilting, we actually wondered if there was something wrong with our simulations,” stated Distinguished Professor of Chemistry and Biochemistry Rommie Amaro, who is the principal investigator on the project. “Once we knew our models were correct, we realized the enormous potential this discovery held. This research could be used to develop methods of keeping the protein locked open so that it would be constantly accessible to antibodies.”
Traditionally, flu vaccines have targeted the head of the HA protein based on still images that showed the protein in a tight formation with little movement. Amaro’s model showed the dynamic nature of the HA protein and revealed a breathing movement that exposed a previously unknown site of immune response, known as an epitope.
This discovery complemented previous work from one of the paper’s co-authors, Ian A. Wilson, Hansen Professor of Structural Biology at The Scripps Research Institute, who had discovered an antibody that was broadly neutralizing—in other words, not strain-specific—and bound to a part of the protein that appeared unexposed. This suggested that the glycoproteins were more dynamic than previously thought, allowing the antibody an opportunity to attach. Simulating the breathing movement of the HA protein established a connection.
NA proteins also showed movement at the atomic level with a head-tilting movement. This provided a key insight to co-authors Julia Lederhofer and Masaru Kanekiyo at the National Institute of Allergy and Infectious Diseases. When they looked at convalescent plasma—that is, plasma from patients recovering from the flu—they found antibodies specifically targeting what is called the “dark side” of NA underneath the head.
Without seeing the movement of NA proteins, it wasn’t clear how the antibodies were accessing the epitope. The simulations Amaro’s lab created showed an incredible range of motion that gave insight into how the epitope was exposed for antibody binding.
The H1N1 simulation Amaro’s team created contains an enormous amount of detail—160 million atoms worth. A simulation of this size and complexity can only run on a few select machines in the world. For this work, the Amaro lab used Titan at Oak Ridge National Lab, formerly one of the largest and fastest computers in the world.
Amaro is making the data available to other researchers who can uncover even more about how the influenza virus moves, grows and evolves. “We’re mainly interested in HA and NA, but there are other proteins, the M2 ion channel, membrane interactions, glycans, so many other possibilities,” Amaro stated.
“This also paves the way for other groups to apply similar methods to other viruses. We’ve modeled SARS-CoV-2 in the past and now H1N1, but there are other flu variants, MERS, RSV, HIV—this is just the beginning.”
More information: Lorenzo Casalino et al, Breathing and Tilting: Mesoscale Simulations Illuminate Influenza Glycoprotein Vulnerabilities, ACS Central Science (2022). DOI: 10.1021/acscentsci.2c00981
Journal information: ACS Central Science
Provided by University of California – San Diego