
by Chiba University
3.H2O belongs to the class of seven-coordinate lanthanide complex compounds considered important for nuclear-fuel processing and magnetic resonance imaging. In this study, researchers developed new force-field parameters for elucidating the structure and dynamics of Ho-(DBM)3.H2O. CMD examinations using the developed force-field revealed that contrary to expectations, the hydrogen bond dynamics of water in the complex are quite like those in bulk water. Credit: Takahiro Ohkubo from Chiba University")
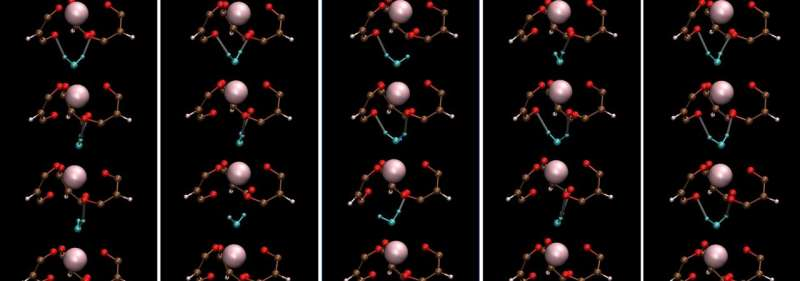
Lanthanide-containing complexes are important compounds for sophisticated nuclear-fuel processing and medical imaging. Moreover, they often have interesting symmetric crystal structures and associated dynamics that render unique properties for practical applications. The seven-coordinate lanthanide complex Ho(III) aqua-tris(dibenzoylmethane) or Ho-(DBM)3·H2O was first reported in the late 1960s.
It has a three-fold symmetric structure with holmium (Ho) at the center of three propeller-shaped dibenzoylmethane (DBM) ligands and a water (H2O) molecule hydrogen-bonded to the ligands. Unfortunately, the understanding of the molecular dynamics (MD) of such lanthanide complexes has been limited due to challenges in describing their interactions using the classical MD framework.
This motivated a team of researchers from the Graduate School of Engineering at Chiba University, led by Associate Professor Takahiro Ohkubo, to elucidate the structure and dynamics of the Ho-(DBM)3·H2O complex. This study was published in Inorganic Chemistry and is co-authored by Associate Professor Hyuma Masu, Professor Keiki Kishikawa, and Associate Professor Michinari Kohri.
“Hydrogen bonds between the water molecule and the ligands surrounding Ho are considered to play an important role in the formation of the symmetrical structure of the novel lanthanide complex. After synthesizing its single crystal and bulk samples, the next logical step was to model this complex to test this hypothesis and understand its structure and dynamics,” explains Dr. Ohkubo.
Considering the shortcomings of existing general force-fields (a functional form used to estimate forces between atoms) in satisfactorily describing the interactions of lanthanide metals such as Ho, the researchers developed new force-field parameters for conducting classical MD simulations of the Ho-(DBM)3·H2O complex. They performed structural optimization and MD steps using ab initio calculations based on the plane-wave pseudopotential method to make training data for force-fields’ development.
Further, the team tuned the force-field parameters for the simulations to reproduce the data obtained from the ab initio calculations. They validated the thus-obtained novel force-field using both the experimental crystalline structure information as well as the theoretical ab initio data. The lattice constant and atomic distances around Ho calculated using the new force-field parameters were found to be in good agreement with the observations of single-crystal X-ray diffraction.
On examining the vibrational properties of water in the Ho-(DBM)3·H2O complex and comparing them to those in bulk liquid water, they observed that the vibrational motion of water in the complex had a characteristic mode.
It originated from stationary rotational motion along the c-axis of Ho-(DBM)3·H2O. Remarkably, the hydrogen bond dynamics of water, including lifetime, in seven-coordinate lanthanide complexes are quite like those in bulk water, except for librational or reciprocating motion. This novel finding is contrary to basic expectations.
In summary, this innovative strategy of developing force-field parameters for classical MD examination unveils the role of water dynamics in complexes such as Ho-(DBM)3·H2O. As Dr. Ohkubo explains, “This approach helped us understand the nature of metal complexes of lanthanides with water and actinide metals with high coordination numbers. In the future, this strategy could possibly pave the way for accurate molecular simulations of any metal complex and prediction of its structure and dynamics.”
More information: Takahiro Ohkubo et al, Molecular Dynamics Studies of the Ho(III) Aqua-tris(dibenzoylmethane) Complex: Role of Water Dynamics, Inorganic Chemistry (2023). DOI: 10.1021/acs.inorgchem.3c01277
Provided by Chiba University