
. DOI: 10.1039/D3SC03598K")
The number of molecules thought to exist is unfathomably large—somewhere between 1050 and 1060 (for comparison, there are only 1022 to 1024 stars in the observable universe). The chemical and pharmaceutical sciences have sought a comprehensive understanding of the fundamental relationships in this vast “chemical compound space” that connects the structure of a given molecule and its properties.
Robert A. DiStasio Jr., associate professor of chemistry and chemical biology in the Cornell University College of Arts and Sciences, and collaborators at the University of Luxembourg and Argonne National Laboratory have conducted an extensive computational study of this space and have introduced a novel concept, called “freedom of design,” that can be used to identify molecules with targeted physical and/or chemical properties. The concept has important implications in the fields of rational molecular design and computational drug discovery.
Their paper, “‘Freedom of Design’ in Chemical Compound Space: Towards Rational in Silico Design of Molecules with Targeted Quantum-Mechanical Properties,” is published in Chemical Science.
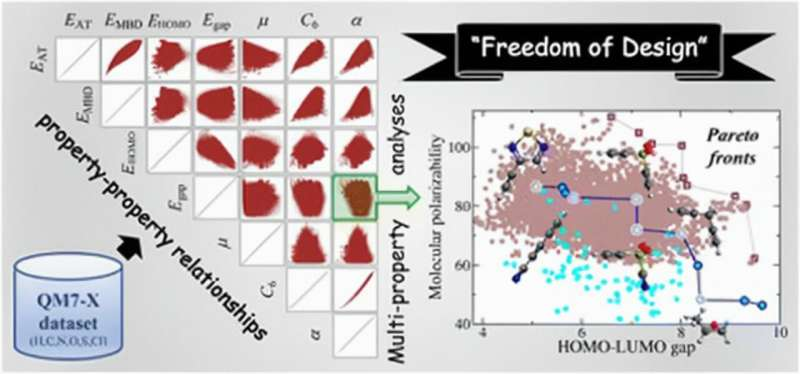
One of the core findings from this international team of researchers was that most molecular properties are only weakly correlated and therefore effectively independent.
“While one might view this as a challenge in the field of rational molecular design, we demonstrate that this finding highlights an intrinsic flexibility—or ‘freedom of design’—that exists in the chemical compound space, wherein there are very few limitations which prevent markedly distinct molecules from sharing multiple important properties,” said DiStasio, a senior author on the paper.
To explore how this flexibility will manifest during the molecular design process, which often involves a “needle-in-a-haystack” search for molecules with a desired set of properties, the authors used “Pareto optimization” to identify potential candidate molecules for building polymeric batteries. In Pareto optimization, changing the molecule would not improve any of its properties without making another property worse.
The search was performed over a collection of molecules that was too large to catalog experimentally, and the results included many unexpected molecules, thereby reflecting the freedom available when designing molecules with targeted properties.
“A potentially interesting next step would be to use these Pareto-optimal structures in conjunction with powerful machine-learning approaches to build reliable multi-objective frameworks for a systematic navigation of hitherto unexplored swaths of chemical compound space,” said Alexandre Tkatchenko, professor of theoretical chemical physics at the University of Luxembourg and the study’s other senior author. “Such a development would enable us to rapidly identify the most promising molecules for next-generation chemical and/or technological applications.”
The insight provided by this work also forms the basis for an overall approach to the rational design of molecules and materials with targeted properties.
“Our understanding of structure-property relationships—the fundamental connections between the structure/composition of molecules and their emergent properties—is at the very heart of chemistry,” DiStasio said. “This work challenges one of the dominant paradigms in the field and begs the question: Which potentially transformative molecules are missed when we only consider modifying the functional groups on a largely fixed molecular scaffold?”
Other collaborators on this work were Leonardo Medrano Sandonas of the University of Luxembourg; Johannes Hoja of the University of Graz in Austria; Cornell doctoral student Brian G. Ernst; and Álvaro Vázquez-Mayagoitia of Argonne National Laboratory.
More information: Leonardo Medrano Sandonas et al, “Freedom of design” in chemical compound space: towards rational in silico design of molecules with targeted quantum-mechanical properties, Chemical Science (2023). DOI: 10.1039/D3SC03598K
Journal information: Chemical Science
Provided by Cornell University