
by FAPESP
. DOI: 10.3390/antibiotics12071202")
Polyalthic acid from copaiba oil is an effective antibacterial and should be used to develop alternative medications that can contribute to the effort to overcome antimicrobial resistance (“superbugs”), according to an article by researchers based in Brazil and the United States published in the journal Antibiotics.
More than 2.8 million antibiotic-resistant infections occur in the US and more than 35,000 people die as a result each year, says a report issued in 2019 by the Centers for Disease Control and Prevention (CDC).
Antimicrobial or antibiotic resistance is when germs (bacteria, fungi) develop the ability to defeat the antibiotics designed to kill them (it does not mean our bodies are resistant to antibiotics). It is expected to become the main global cause of death by 2050.
The crisis is due to improper prescribing of antibiotics, intense use of these drugs in agriculture, and overuse of a small number since the leading pharmaceutical companies decided to abandon the development of antibiotics owing to high cost and low return on investment.
In this context, resorting to plants as a source of novel drugs has proved a promising alternative.
To stimulate knowledge production in this field, researchers in Brazil at the University of São Paulo’s Ribeirão Preto School of Pharmaceutical Sciences (FCFRP-USP) and São Carlos Institute of Physics (IFSC-USP), in collaboration with colleagues at the University of Franca (UNIFRAN), also in Brazil, and the College of Pharmacy and Health Sciences at Western New England University (WNE) in the US, investigated copaiba oil, derived from Copaifera trees and traditionally used in the Amazon region as a natural remedy for its wound-healing, anti-inflammatory and antimicrobial properties. Its main constituents are diterpenes (20%), including polyalthic acid, and sesquiterpenes (80%). Both groups of compounds are anti-inflammatory and antimicrobial.
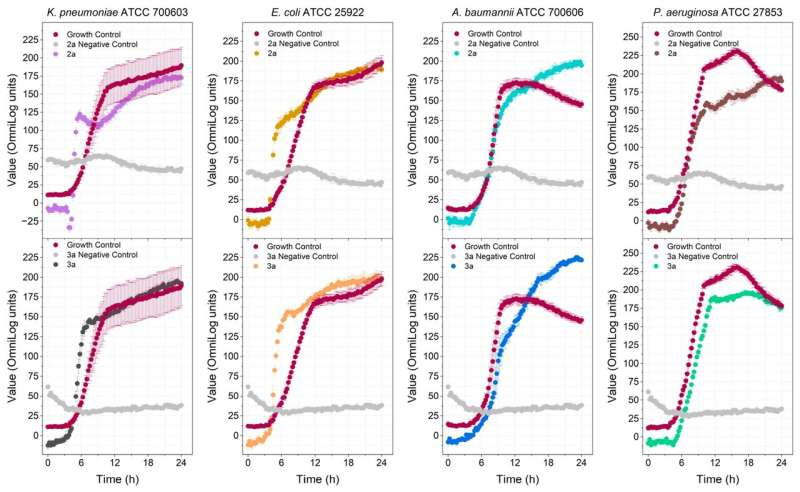
The researchers synthesized four polyalthic acid analogs with structural modifications to make them more active against pathogens, and investigated their efficacy against biofilms of Staphylococcus epidermidis, a bacterium that causes skin and digestive tract infections, and against several Gram-positive bacteria (Enterococcus faecalis, Enterococcus faecium, S. epidermidis and Staphylococcus aureus). They also determined the minimum dosage required to inhibit planktonic (free-floating) bacteria.
. Credit: Antibiotics (2023). DOI: 10.3390/antibiotics12071202")
Activity tests and comparisons with the original polyalthic acid and the drug most prescribed by physicians showed that the analogs developed by the researchers eradicated S. epidermidis, and were active against all the Gram-positive bacteria tested. Although they were less active than the prescribed drug, the results reinforced the importance of additional in vitro and in vivo testing of the substance.
“The advantage of studying polyalthic acid is that previous research has shown that some terpenes don’t lose their activity, and their continuous use therefore doesn’t make bacteria develop resistance,” said Cássia Suemi Mizuno, a researcher at WNE and last author of the article.
The analogs were found to be safe in an analysis of hemolytic activity, i.e. their ability to destroy red blood cells.
Next steps
“Our research is an important contribution to efforts to beat antimicrobial resistance and serves as a foundation on which other groups can made further progress,” Mizuno said.
Next steps will include producing more derivatives with other parts of the polyalthic acid molecule, improving their activity and pursuing prospective partners in the pharmaceutical industry for more research, she added.
Investment in copaiba oil extraction in the Amazon will be needed, as will the recruitment of forest dwellers who are familiar with the native vegetation and can identify the species with the highest level of polyalthic acid content (Copaifera reticulata Ducke).
“It should be stressed that we don’t destroy any trees in our research. Extraction of copaiba oil is like rubber tapping. You just make a groove in the bark of the tree trunk,” Mizuno said.
More information: Marcela Argentin et al, Synthesis and Antibacterial Activity of Polyalthic Acid Analogs, Antibiotics (2023). DOI: 10.3390/antibiotics12071202
Provided by FAPESP




. DOI: 10.1039/D3SC01827J")
. DOI: 10.1039/D3SC01827J")
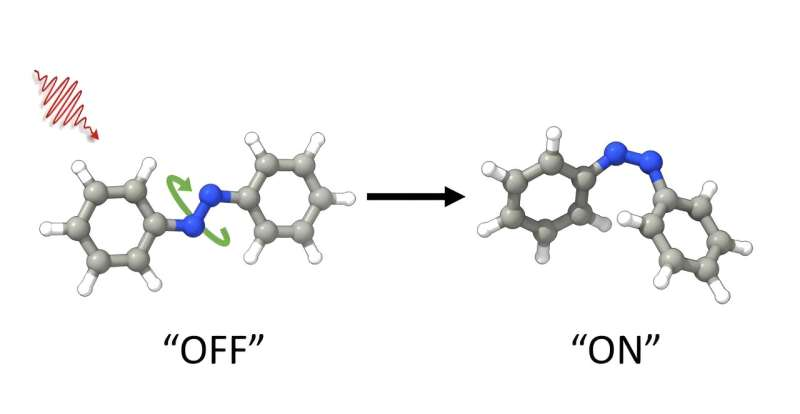

 and a photosensitizer are confined in a molecular cage. The confined space of the cage fits only an E-form of azobenzene but not the Z-form of azobenzene, which is expulsed from the cage. Credit: Jonathan R. Church")
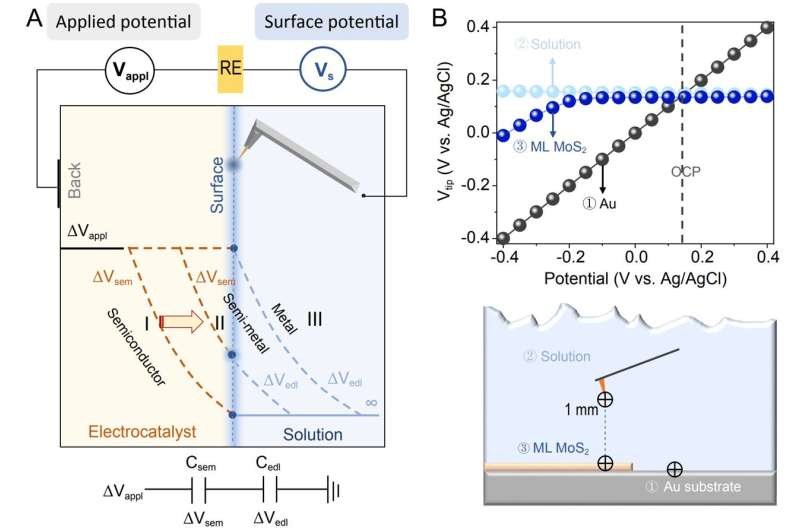
 a schematic of in-situ surface potential measurement set-up and potential drops acrossthe electrode–electrolyte interface for metal, semi-metal and semiconductor respectively. (b) surface potential values of the basal plane of ml molybdenum disulfide, au and solution were measured with a tip in 0.1 m K2SO4. Credit: Science China Press")
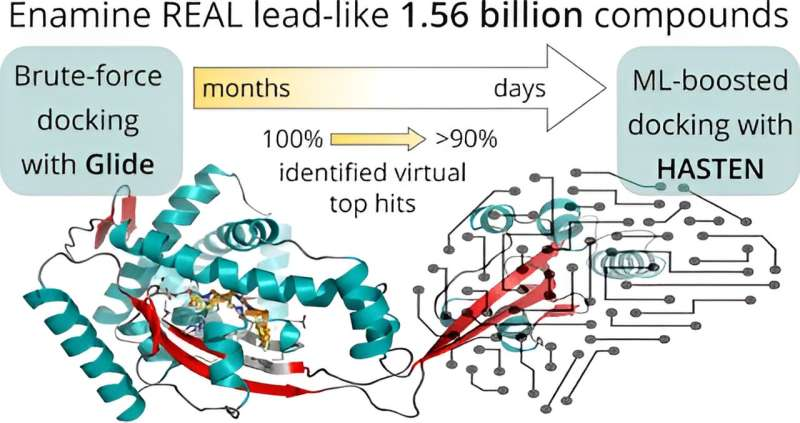
. DOI: 10.1021/acs.jcim.3c01239")




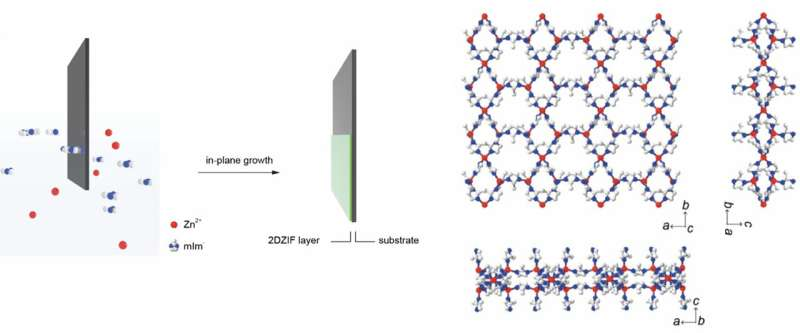
. Right: Crystal structure of 2d ZIF where white, blue, and red atoms represent carbon, nitrogen, and zinc atoms. Credit: Qi Liu, EPFL.")
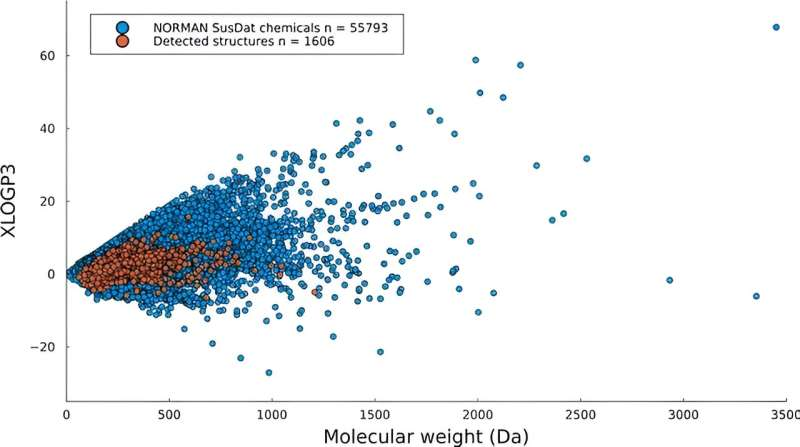
, overlaid on top of a distribution of the NORMAN SusDat database chemicals (blue), based on their molecular weights (x-axis) and XLOGP3 values (y-axis). Credit: HIMS / EST")