

by ETH Zurich

Researchers from ETH Zurich and the University of Geneva have developed a new method that allows them to observe chemical reactions taking place in liquids at extremely high temporal resolution. This means they can examine how molecules change within just a few femtoseconds—in other words, within a few quadrillionths of a second. The method is based on earlier work done by the same group of researchers led by Hans Jakob Wörner, Professor of Physical Chemistry at ETH Zurich. That work yielded similar results for reactions that take place in gas environments.
To expand their X-ray spectroscopy observations to liquids, the researchers had to design an apparatus capable of producing a liquid jet with a diameter of less than one micrometer in a vacuum. This was essential because if the jet were any wider, it would absorb some of the X-rays used to measure it. The work is published in the journal Nature.
Molecular pioneer in biochemistry
Using the new method, the researchers were able to gain insights into the processes that led to the emergence of life on Earth. Many scientists assume that urea played a pivotal role here. It is one of the simplest molecules containing both carbon and nitrogen. What’s more, it’s highly likely that urea was present even when the Earth was very young, something that was also suggested by a famous experiment done in the 1950s: American scientist Stanley Miller concocted a mixture of those gases believed to have made up the planet’s primordial atmosphere and exposed it to the conditions of a thunderstorm. This produced a series of molecules, one of which was urea.
According to current theories, the urea could have become enriched in warm puddles—commonly called primordial soup—on the then lifeless Earth. As the water in this soup evaporated, the concentration of urea increased. Through exposure to ionizing radiation such as cosmic rays, it’s possible that this concentrated urea produced malonic acid over multiple synthesis steps. In turn, this may have created the building blocks of RNA and DNA.
Why this exact reaction took place
Using their new method, the researchers from ETH Zurich and the University of Geneva investigated the first step in this long series of chemical reactions to find out how a concentrated urea solution behaves when exposed to ionizing radiation.
It’s important to know that the urea molecules in a concentrated urea solution group themselves into pairs, or what are known as dimers. As the researchers have now been able to show, ionizing radiation causes a hydrogen atom within each of these dimers to move from one urea molecule to the other. This turns one urea molecule into a protonated urea molecule, and the other into a urea radical. The latter is highly chemically reactive—so reactive, in fact, that it’s very likely to react with other molecules, thereby also forming malonic acid.
The researchers also managed to show that this transfer of a hydrogen atom happens extremely quickly, taking only around 150 femtoseconds, or 150 quadrillionths of a second. “That’s so fast that this reaction preempts all other reactions that might theoretically also take place,” Wörner says. “This explains why concentrated urea solutions produce urea radicals rather than hosting other reactions that would produce other molecules.”
Reactions in liquids are highly relevant
In the future, Wörner and his colleagues want to examine the next steps that lead to the formation of malonic acid. They hope this will help them to understand the origins of life on Earth.
As for their new method, it can also generally be used to examine the precise sequence of chemical reactions in liquids. “A whole host of important chemical reactions take place in liquids—not just all biochemical processes in the human body, but also a great many chemical syntheses relevant to industry,” Wörner says. “This is why it’s so important that we have now expanded the scope of X-ray spectroscopy at high temporal resolution to include reactions in liquids.”
The researchers from ETH Zurich and the University of Geneva were assisted in this work by colleagues from Deutsches Elektronen-Synchrotron DESY in Hamburg, who performed calculations required to interpret measurement data.
More information: Hans Jakob Wörner, Femtosecond Proton Transfer in Urea Solutions Probed by X-ray Spectroscopy, Nature (2023). DOI: 10.1038/s41586-023-06182-6. www.nature.com/articles/s41586-023-06182-6
Journal information: Nature
Provided by ETH Zurich
. Credit: Diamond Light Source Ltd")


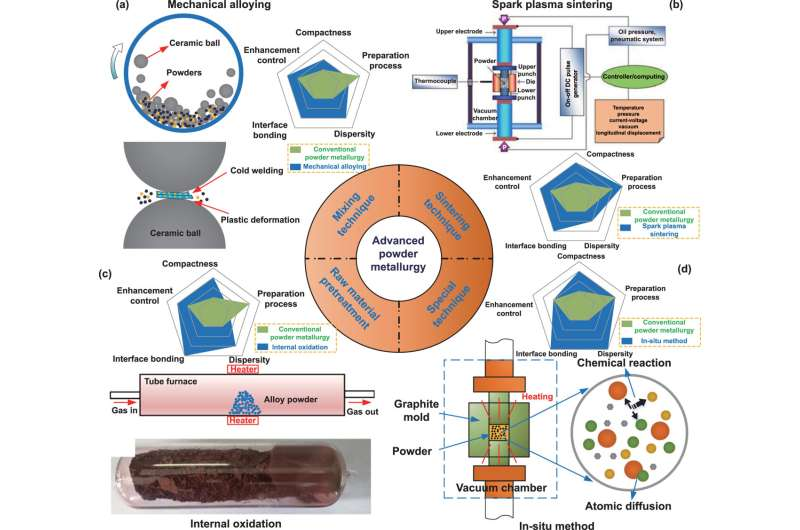
 Mechanical alloying, (b) spark plasma sintering. Reproduced from [29]. CC BY 3.0. (c) Internal oxidation. Reprinted from [30], Copyright (2019), with permission from Elsevier.(d) In-situ processing. Credit: International Journal of Extreme Manufacturing (2023). DOI: 10.1088/2631-7990/acdb0b")
")

, and graduate students Xuanxin Hu(middle) and Nuohao Liu (right) have developed new criteria for determining whether shearbands are beneficial or harmful to certain crystalline materials. Credit: University of Wisconsin–Madison")


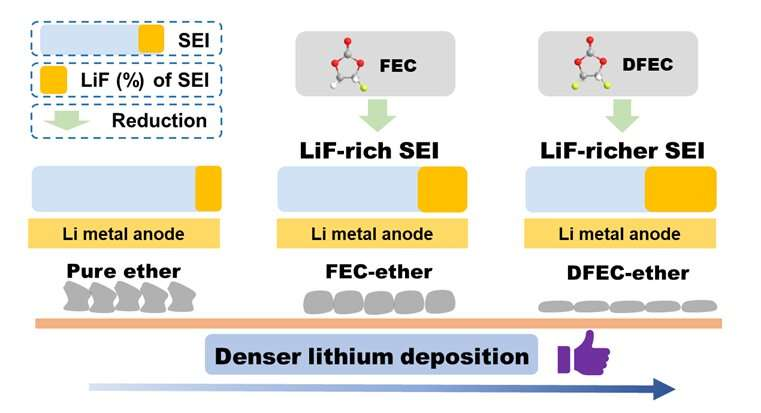
 and affection on Li deposition in ether electrolyte. Credit: Science China Press")
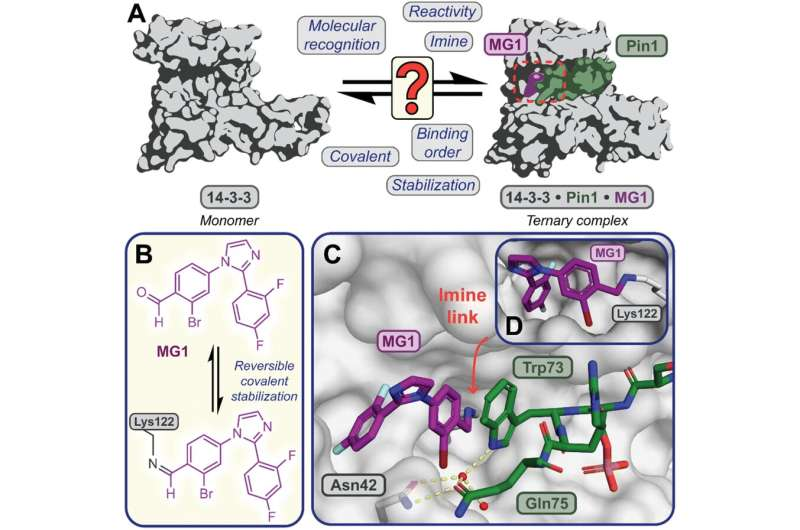
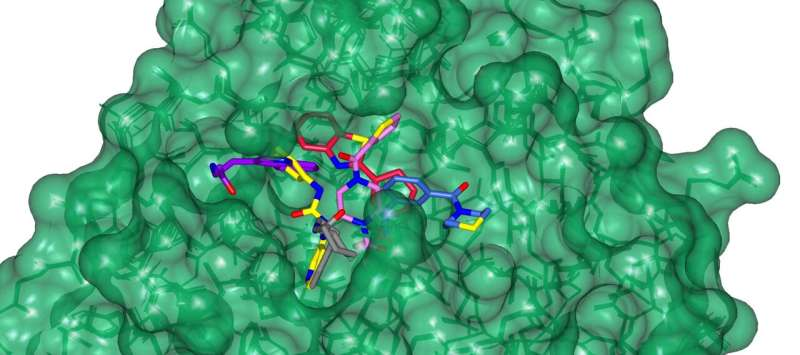
 Illustrates the limited understanding of MG1 induced complex stabilization. (B) Schematic representation of MG1 ligation. (C) Enlarged view of the 14-3-3/Pin1/MG1 interface . (D) MG1 covalently bound to Lys122 of 14-3-3 by aldimine bonding. Credit: Chemical Science (2023). DOI: 10.1039/D3SC01732J")


